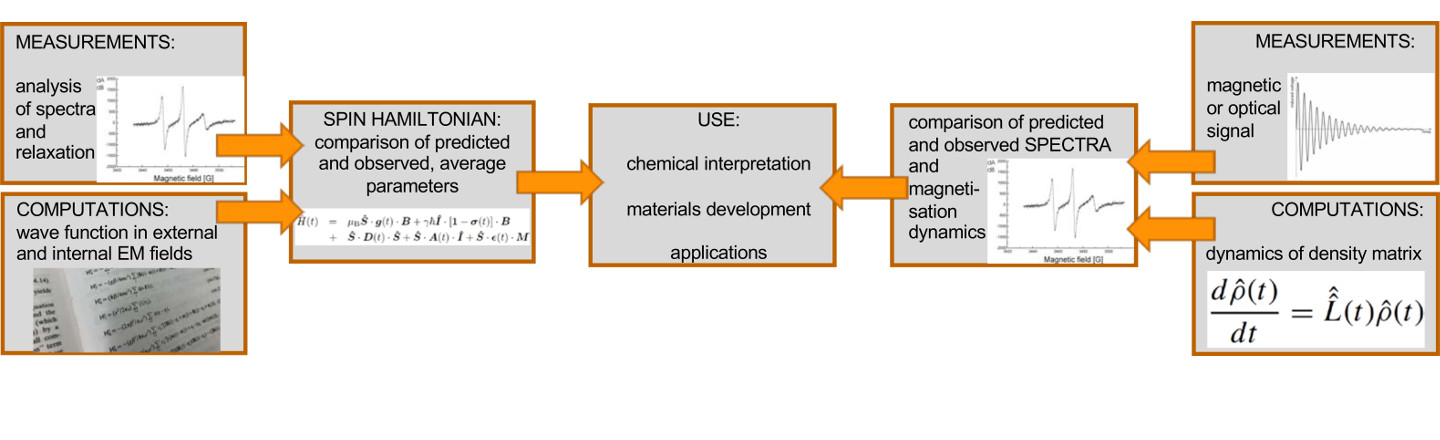
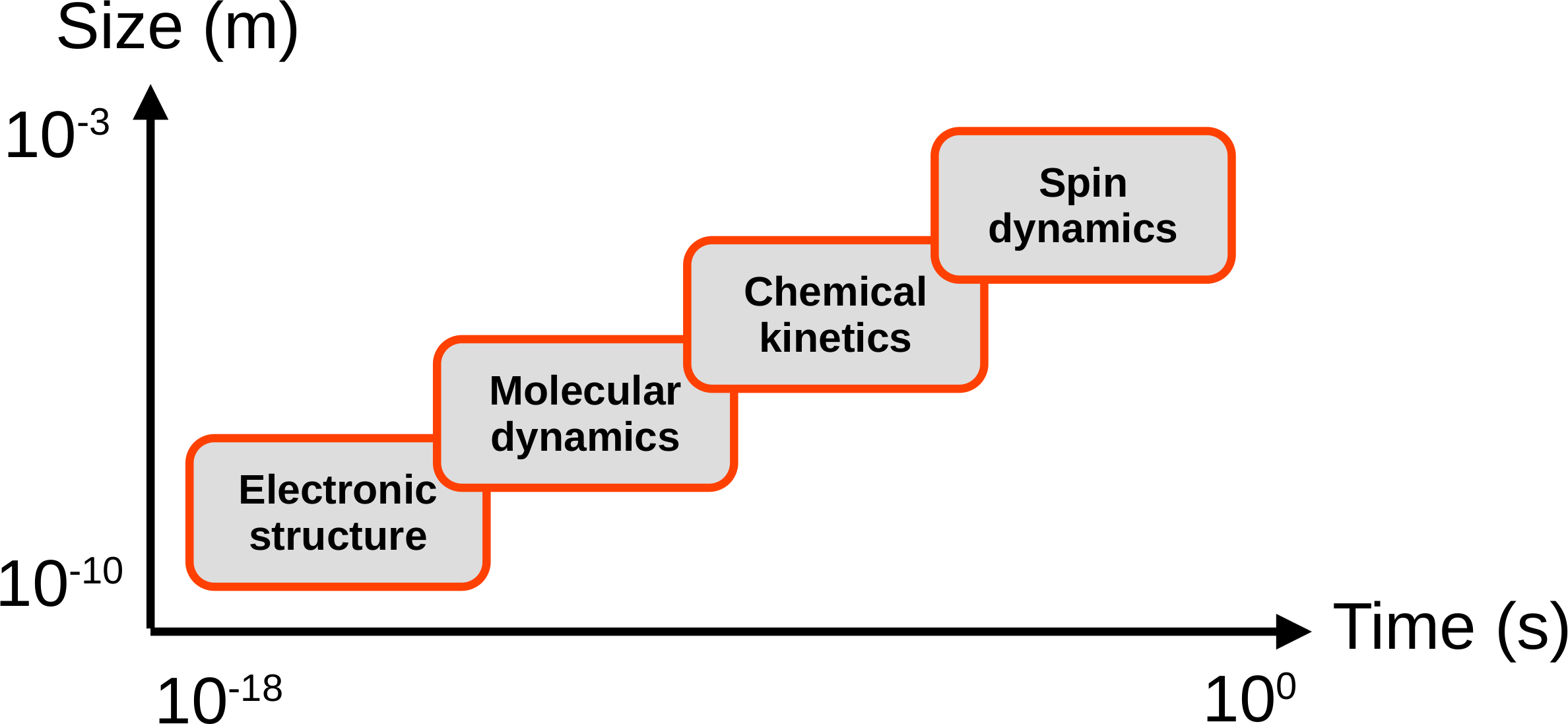
An emerging paradigm in theoretical molecular and materials sciences is computational spectroscopy, which in the case of magnetic resonance spectroscopy means computing directly the primary experimental observables, the time-dependent magnetization or the resulting spectrum. This is to be compared with the traditional way in which comparison with experiment is done at the level of NMR spectral parameters or relaxation times, which themselves result from applying theoretical models to the measurement data. We realise computational spectroscopy by multiscale modelling of
atomic motion within the material via molecular dynamics simulations;
electronic interactions of the spins at each set of atomic positions via advanced quantum-chemical calculation of a time series of spin Hamiltonians,
chemical exchange between different environments of the spin system, and
the resulting time dependence of the nuclear and electron spin density matrices obtained by integrating the Liouville-von Neumann equation.
This procedure enables detailed, microscopic interpretation and development of relaxation and polarisation transfer processes of relevance to modern magnetic resonance.
Movie 1: Trajectory of an individual Rb-Xe pair in a spin-exchange optical pumping simulation. The bond length (green) of the van der Waals complex and the polarisation (blue) of the 129Xe nucleus, transferred from the optically polarised Rb electron, are shown. The depicted complex is disturbed by several intruding gas molecules, which change the dynamics of the complex and almost lead (around 3500 x 50 fs) to break-up. [Rantaharju, Hanni and Vaara, Phys. Rev. A 102, 032813 (2020)].
The currently investigated topics include realistic modelling of the SEOP polarisation transfer to xenon, first-principles relaxation of gaseous xenon, accurate spin Hamiltonian of the NV- centres in diamond, molecular and spin dynamics of xenon in paramagnetic cage structures, spin dynamics of Hyper-CEST experiments on xenon in biosensor structures and (in collaboration with the Kantola group) polarisation transfer simulations in SABRE, as well as development of the Spinguin spin dynamics simulation package.
Movie 2 (a): Molecular dynamics simulation of the Xe@CrA biosensor structure in aqueous solution. The detailed atomic dynamics produces a fluctuating dipole-dipole interaction between the spin of the 129Xe guest and the 1H nuclei of the CrA host cage. (b): When the guest is hyperpolarised by spin-exchange optical pumping, and then chemically exchanged from the solution to the cage, the dipole-dipole fluctuations lead to relaxation of the 129Xe spin, spin polarisation-induced nuclear Overhauser enhancement (SPINOE) of the host proton magnetisation and spin diffusion between the different proton groups of the cage. [Hilla and Vaara, Phys. Chem. Chem. Phys. 27, 10979 (2025)].